体外诊断试剂产品注册技术审评报告
产品中文名称:人类10基因突变联合检测试剂盒 (可逆末端终止测序法)
产品管理类别:三类6840
申请人名称: 厦门艾德生物医药科技股份有限公司
国家食品药品监督管理总局
医疗器械技术审评中心
基本信息
一、申请人名称:厦门艾德生物医药科技股份有限公司
二、申请人住所:厦门市海沧区鼎山路39号
三、生产地址:厦门市海沧区鼎山路39号
产品审评摘要
一、产品概述
(一)产品主要组成成分
表1 试剂盒主要组成成分
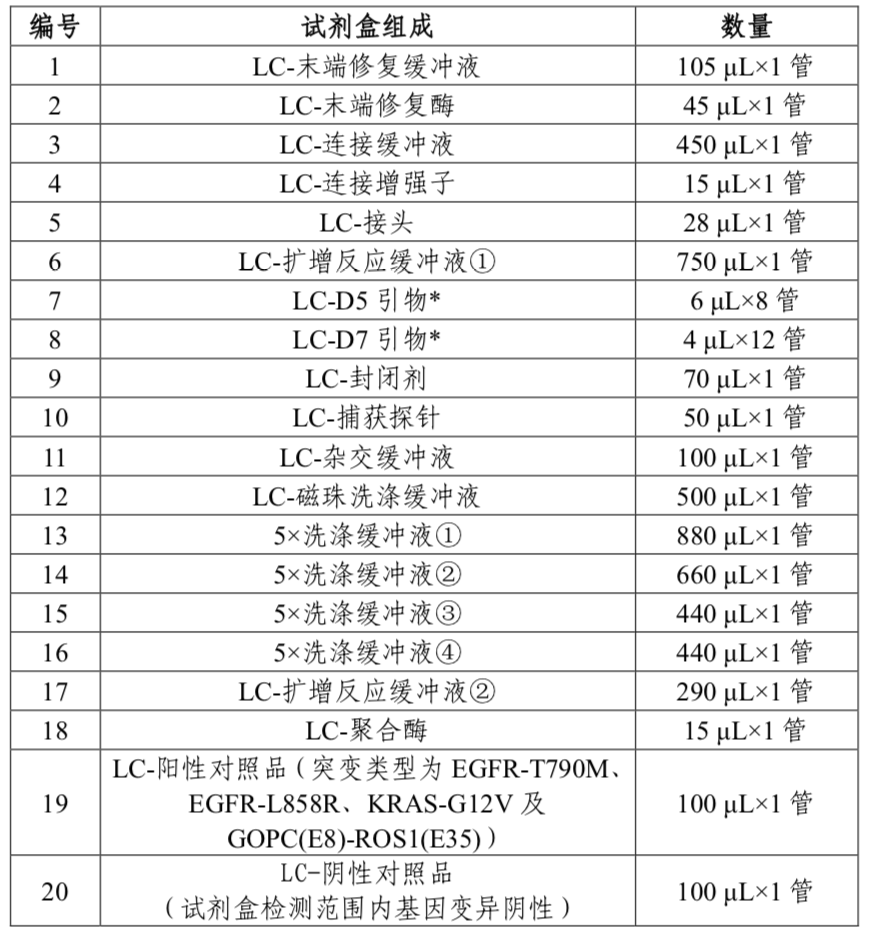
具体组成成分、配套试剂及软件见说明书。
(二)产品预期用途
本试剂盒用于定性检测非小细胞肺癌(NSCLC)、结直肠癌(CRC)患者经中性福尔马林固定的石蜡包埋(FFPE)的组织样本中EGFR/ALK/ROS1/RET/KRAS/NRAS/PIK3CA/BRAF/HER2/MET基因变异。其中,针对NSCLC,EGFR基因中:19号外显子缺失(19del)、L858R点突变用于吉非替尼片的伴随诊断检测,T790M点突变用于甲磺酸奥希替尼片的伴随诊断检测;ALK基因重排(融合)和ROS1基因重排(融合)用于克唑替尼胶囊的伴随诊断检测;针对CRC,KRAS基因野生型用于西妥昔单抗注射液的伴随诊断检测;如表2所示。
表2 伴随诊断用途的基因变异类型及相应的靶向药物

表3中为本试剂盒可以检出,但未经伴随诊断验证的基因变异类型。
表3 未经伴随诊断用途验证的基因变异类型
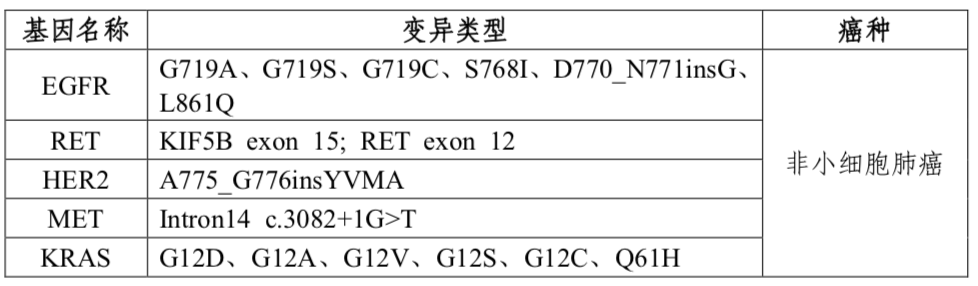
其检测结果仅供临床参考,不应作为患者个体化治疗的唯一依据。临床医生应结合患者病情及其他实验室检测指标等因素对检测结果进行综合判断。
(三)产品包装规格
24测试/盒
(四)产品检验原理
本试剂盒通过构建样本DNA测序文库,并使用特异探针对文库进行目标区域捕获富集,捕获后的文库通过高通量测序,可实现多个基因多个突变的一次性检测。
首先,以FFPE样本提取的DNA为材料,进行DNA片段化处理,并使用磁珠对DNA进行片段选择,然后对所得片段进行末端修复和加A处理,使用DNA连接酶将接头连接到DNA模板的两端,再使用带有标签序列(Index)的引物和聚合酶进行PCR扩增得到扩增文库;扩增文库与带有生物素标记的寡核苷酸探针进行液相杂交,并用链霉素包被的磁珠对与探针结合的文库进行捕获富集,最后经过PCR扩增得到捕获文库。捕获文库采用基因测序仪(型号:NextSeq CN500,杭州贝瑞和康基因诊断技术有限公司生产,注册证号:国械注准20153400460)进行高通量测序。对于测序数据,采用生物信息学软件判读10种基因中是否存在来自肿瘤的变异。
二、临床前研究摘要
(一)主要原材料
1.主要原材料的选择
该试剂盒主要原材料包括:捕获探针、末端修复酶、连接酶、DNA聚合酶、对照品等,这些原材料均为外购方式获得,捕获探针为申请人自行设计后由专业的合成公司合成。申请人选择有资质的供应商提供的原料,通过功能性试验,筛选出最佳原材料和供应商。制定了各主要原材料质量标准并经检验合格。
2.企业参考品和质控品设置情况
企业参考品包括阳性参考品、阴性参考品、检测限参考品和重复性(精密度)参考品。其中:
阳性参考品共18份,包括该产品可检出的所有突变或融合类型,均为DNA样本。其中8份来自临床样本,5份来自混合的细胞系参考品(由自行购买的不同突变或融合类型的15个细胞系参考品混合而成),5份来自混合人工合成DNA的人类细胞系参考品(一共含18个不同突变或融合类型)。所有参考品均用Sanger测序法和数字PCR方法验证。
阴性参考品共8份,包括1份大肠杆菌样本,7份试剂检测范围内基因变异阴性的临床样本。所有样本均用Sanger测序法和数字PCR方法验证为相关突变阴性。
检测限参考品共10份,包括该产品可检出的所有突变或融合类型。其中5份由混合阳性细胞系和试剂检测范围内基因变异阴性的细胞系DNA稀释获得,5份由人工合成DNA和试剂检测范围内基因变异阴性的细胞系样本DNA稀释获得。10份检测限参考品稀释后的突变类型和重排(融合)类型变异比例为1%。
重复性参考品总共15份,包括该产品可检出的所有突变或融合类型。其中7份由混合阳性细胞系和试剂检测范围内基因变异阴性的细胞系DNA稀释获得,7份由人工合成DNA和试剂检测范围内基因变异阴性的细胞系样本DNA稀释获得,1份为试剂检测范围内基因变异阴性的细胞系样本DNA。12份弱阳性参考品突变比例为1~5%,2份强阳性参考品突变比例为15%,另采用试剂检测范围内基因变异阴性的细胞系制成1份阴性参考品。
该产品的对照品来源于细胞系样本的DNA,其中阳性对照品包含EGFR/KRAS基因突变阳性和ROS1基因融合,阴性对照品为试剂检测范围内10种基因变异阴性,用于检测过程中试剂和仪器的质量控制。
(二)生产工艺及反应体系研究
申请人通过对试剂主要生产工艺的研究,确定了最佳生产工艺。
申请人通过使用初步确定的配方进行反应体系配制,对反应体系中的样本核酸提取试剂、末端修复试剂用量、连接试剂用量、接头用量、LC-D7/D5引物用量、杂交文库混合数量、杂交捕获文库用量、封闭剂用量、捕获探针用量、杂交液用量、磁珠用量、杂交时间、扩增反应条件等进行筛选和优化,通过功能性试验,最终确定了最佳的反应体系。
(三)分析性能评估
分析性能评估内容包括阴/阳性参考品符合率、最低检出限、分析特异性和干扰物质、重复性、肿瘤组织样本要求研究、核酸提取纯化配套试剂组合性能研究等。
在阴/阳性参考品符合率试验中,申请人采用18份阳性参考品和8份阴性参考品对3批成品试剂盒(P215071301Z、P215071501Z、P215071701Z)进行了检测验证,检测结果均为预期的突变型,说明阳性参考品符合率和阴性参考品符合率均为100%。同时选取21个阳性临床样本在3批成品试剂盒(06018010501Z、06018032901Z、06018052101Z)上分别进行检测,结果均能正确检出。
在最低检出限试验中,申请人采用10份检测限参考品对3批成品试剂盒(P215071301Z、P215071501Z、P215071701Z)进行了检测验证,结果均能正确检出。同时选取4份经过数字PCR定量的阳性临床样本(EGFR/ALK/HER2基因变异阳性,含有点突变、插入缺失突变和基因重排),用基因变异阴性的临床样本配制8级突变频率梯度共32个样本,每个样本分别在3种建库起始量(10 ng、30 ng和50 ng)下进行了6次重复检测,确定了样本的检测限为可检测30ng DNA中频率低至1%的突变(含有点突变、插入缺失突变和基因重排)。
取15份经过数字PCR定量的阳性临床样本(EGFR/ALK/ROS1/HER2/RET/KRAS/NRAS/PIK3CA/BRAF基因变异阳性,含有点突变、插入缺失突变和基因重排),用试剂检测范围内基因变异阴性的临床样本配制成1%变异频率,在30ng建库起始量下,用3批成品试剂盒(06018010501Z、06018032901Z、06018052101Z)对各样本进行了20次重复检测验证,最终确定该产品不低于95%检出率时,可检测30ng DNA中频率低至1%的突变(含有点突变、插入缺失突变和基因重排)。
在分析特异性试验中,申请人采用试剂盒检测范围内的基因变异阴性的临床样本DNA,进行不同建库起始量的检测,检测结果均为阴性,说明产品对不同起始量的阴性临床样本DNA具有良好的耐受性。对于试剂盒检测范围内的基因变异阳性参考品,本试剂盒检测结果均为相应型别阳性,没有错检或漏检,说明本试剂盒能准确检测目标区域的碱基突变状态,检测的变异类型间不存在交叉反应。本试剂盒能对非人类基因组(大肠杆菌和肺炎链球菌)DNA进行正常建库,但不能对目标区域序列进行捕获富集,说明本试剂盒与非人类基因组DNA无交叉反应。
在干扰物质试验中,申请人在阳性临床样本和阴性临床样本中分别加入如下干扰物质:坏死组织(等体积)、乙醇(21.7 mmol/L)、二甲苯(35 mmol/L)、蛋白酶K(0.08 mg/mL)然后进行检测,样本检测结果均与预期一致,说明这些干扰物质在不高于上述浓度的情况下不会对该产品的结果产生影响。
在重复性试验中,申请人采用重复性参考品15份在3批成品试剂盒(P215071301Z、P215071501Z、P215071701Z)上分别完成20次重复检测,检测结果一致。同时选取了21份临床样本,在3批成品试剂盒(06018010501Z、06018032901Z、06018052101Z)上分别完成20次重复检测,检测结果一致。
针对核酸提取纯化步骤,申请人采用临床样本,平行比较了2种核酸提取试剂盒的提取效果,根据与该产品的组合性能研究结果,确定了1种核酸提取试剂作为样本提取的推荐试剂盒。
在肿瘤组织细胞含量研究中,申请人对不同肿瘤细胞占比对检测结果的影响进行了研究。结果表明,肿瘤细胞占比5%以上的组织样本均可检出。结合临床样本的多样性和复杂性,建议组织样本肿瘤细胞占比≥20%。
申请人提供了3批产品(06216070401X,06216071101X,06216071801X)在其适用机型上的性能评估资料,结果均符合要求。
(四)阳性判断值
申请人首先采用少量临床样本进行了初步确定,然后采用不同基因不同变异类型的阳性临床样本进行阳性判断值验证,最终确定了阳性判断值标准。
申请人共采用了38个样本(16个商业化参考品及22个临床样本;包含了点突变、插入/缺失和融合三种类型)进行检测,分别统计其检出情况。通过ROC曲线方式确认采用突变频率0.4%可以有效检出突变。并对样本原始数据分别随机抽取测序深度为15000×、10000×、7500×、5000×,确定样品测序平均原始深度要求为10000×,并确定了位点有效深度的最低要求为500×,突变绝对拷贝数应不低于2,选择链平衡性0.1~0.9作为阳性判断值的要求(融合不适用)。
阳性判断值验证:采用10份阴性、10份灵敏度参考品、以及经数字PCR验证并稀释至1%的15例FFPE样本(包含不同基因不同区段的变异类型),分别测试阳性符合率、阴性符合率,结果表明,阳性符合率、阴性符合率都100%吻合,总体验证结果符合要求。
通过上述实验,最终确定该产品使用配套软件进行数据分析时的阳性判断值为:
1.突变比例不低于0.4%;
2.测序有效深度不低于500×;
3.突变绝对拷贝数不低于2;
4.链平衡性介于0.1~0.9之间(融合不适用)。
(五)稳定性研究
申请人对该产品实时稳定性、运输稳定性、冻融稳定性、开瓶稳定性进行了研究,确定了在各种条件下本产品的有效保存时间。同时对石蜡样本稳定性、核酸(DNA)溶液稳定性、文库稳定性等进行了研究,确定了检测过程中各种样本类型的有效保存时间。
实时稳定性研究:采用三批次试剂盒(P215090202Z、P215090603Z、P215100801Z)储存于-20±5℃条件下,分别在0、3月、6月和8月对物理性能、准确度、特异性、检测限和精密度进行考察。结果显示,各项性能指标均符合要求,确定产品在-20±5℃条件下,可稳定保存6个月。
运输稳定性研究:采用三批试剂盒(P215090202Z、P215090603Z、P215100801Z)按运输要求完成运输试验,然后按要求储存至效期末,在运输前/后、储存至效期末时分别对物理性能、准确度、特异性、检测限和精密度进行考察。结果显示,各项性能指标均符合要求,确定了该产品的运输条件。
冻融稳定性研究:采用三批次试剂盒(P215090202Z、P215090603Z、P215100801Z)反复冻融10次,分别在反复冻融第5次和10次后对物理性能、准确度、特异性、检测限和精密度进行考察。结果显示,各项性能指标均符合要求。考虑到临床实际运用,为保证检测结果的准确性,建议冻融次数不超过5次。
开瓶稳定性研究:采用三批次试剂盒(P215090202Z、P215090603Z、P215100801Z)解冻后,拆开包装,将末端修复缓冲液、末端修复酶、连接缓冲液、连接增强子、接头、扩增反应缓冲液、LC-D5引物、LC-D7引物、封闭剂、捕获探针、杂交缓冲液、磁珠洗涤缓冲液、5×洗涤缓冲液、聚合酶、对照品等各组分震荡混匀、瞬时离心,在超净工作台中依次开盖后再盖紧,放置5分钟后置于-20±5℃存储(模拟使用过程),分别在第3和6个月对物理性能、准确度、特异性、检测限和精密度进行考察。结果显示,开瓶后的试剂盒在-20±5℃保存6个月,各项性能指标均满足要求。
申请人对石蜡包埋样本稳定性、核酸(DNA)样本冻融稳定性也进行了研究。研究确认石蜡组织样本保存期限不超过18个月;核酸(DNA)样本置于-20±5℃保存期限可达到8个月。
三、临床评价摘要
本产品用于定性检测非小细胞肺癌(NSCLC)、结直肠癌(CRC)患者经中性福尔马林固定的石蜡包埋(FFPE)的组织样本中EGFR/ALK/ROS1/RET/KRAS/NRAS/PIK3CA/BRAF/HER2/MET基因变异。其中,针对NSCLC,EGFR基因中:19号外显子缺失(19del)、L858R点突变用于吉非替尼片的伴随诊断检测,T790M点突变用于甲磺酸奥希替尼片的伴随诊断检测;ALK基因重排(融合)和ROS1基因重排(融合)用于克唑替尼胶囊的伴随诊断检测;针对CRC,KRAS基因野生型用于西妥昔单抗注射液的伴随诊断检测。
(—)比较研究
申请人在福建医科大学附属协和医院、第四军医大学附属唐都医院、福建省立医院、首都医科大学附属北京胸科医院共4家临床试验机构完成了临床试验。采用考核试剂与组织检测的金标准Sanger测序法对临床样本进行比较研究,验证本产品的临床性能。入组样本为非小细胞肺癌样本1248例、结直肠癌样本295例和干扰样本20例,共计1563例有效样本。样本类型均为石蜡包埋组织。考核试剂与对比试剂检测结果不一致的样本采用PCR方法进行验证。
本临床试验在1248例肺癌样本中共检出863例突变阳性样本,阳性率为69.15%。包括EGFR基因中335例L858R突变阳性、270例Exon19 deletion突变阳性、12例T790M突变阳性、14例L861Q突变阳性、6例S768I突变阳性、8例G719A突变阳性、9例G719S突变阳性;KRAS基因中33例G12D突变阳性、33例G12C突变阳性、29例G12V突变阳性、7例G12A突变阳性、4例G12S突变阳性、2例Q61H突变阳性;NRAS基因中G12D和Q61K突变阳性各1例;BRAF基因中13例V600E突变阳性;PIK3CA基因中17例H1047R突变阳性;MET基因中检出申报类型和非申报类型共20例MET Exon14 skipping阳性突变;HER2基因中24例A775_G776insYVMA突变阳性;ALK基因中检出申报类型和非申报类型共85例ALK基因重排(融合)阳性; ROS1基因中检出申报类型和非申报类型共13例ROS1基因重排(融合)阳性;RET基因中检出申报类型和非申报类型17例RET基因重排(融合)阳性。
与对比方法的研究结果显示,考核试剂的定性检测结果阳性符合率为98.99%(95%CI 98.02%~99.56%),阴性符合率为82.49%(95%CI 78.69%~85.87%),总符合率为92.95%(95%CI 91.38%~94.31%)。进行Kappa一致性检验,Kappa值=0.8429,P<0.001,显示二者具有良好的检测一致性。
在1248例肺癌样本中,两种方法检测结果不一致或不完全一致共有109例。109例样本的第三方验证结果中,有92例与考核试剂检测结果一致,有14例与对照方法检测结果一致,2例(双突变样本)与双方检测结果均不一致,1例因特殊情况未进行验证。
本临床试验在295例结直肠癌样本中共检出111例突变阳性样本,阳性率为37.63%。包括KRAS基因中45例G12D突变阳性、10例G12C突变阳性、17例G12V突变阳性、3例G12A突变阳性、3例G12S突变阳性;NRAS基因中8例G12D突变阳性、3例Q61K突变阳性、2例Q61R突变阳性;BRAF基因中13例V600E突变阳性;PIK3CA基因中9例H1047R突变阳性;1例EGFR基因G719S突变阳性;1例ROS1基因重排(融合)阳性。
与对比方法的研究结果显示,考核试剂的定性检测结果阳性符合率为100%(95%CI 96.38%~100%),阴性符合率为94.36%(95%CI 90.13%~97.15%),总符合率为96.27%(95%CI 93.43%~98.12%)。进行Kappa一致性检验,Kappa值=0.9190,P<0.001,显示二者具有良好的检测一致性。
在295例结直肠癌样本中,两种方法检测结果不一致或不完全一致共有11例。11例样本的第三方验证结果中,有7例与考核试剂检测结果一致,有3例与对照方法检测结果一致,1例(双突变样本)与双方检测结果均不一致。
(二)伴随诊断比较研究
申请人在山西省肿瘤医院进行考核试剂与伴随诊断用途试剂的比较研究。
1.1. EGFR基因与已上市伴随诊断试剂盒的比较研究
EGFR基因突变对比试剂采用凯杰公司therascreen®EGFR RGQ PCR Kit试剂盒。本次对比研究共纳入220例福尔马林固定,石蜡包埋的晚期非小细胞肺癌组织样本。考核试剂在83例样本中检出阳性(其中19Del 41例、L858R 39例、T790M 3例、L861Q3例、S768I 1例)。
考核试剂在220例样本中有6例样本定性检测结果与对照试剂不一致。其中5例为考核试剂检出阳性(其测序丰度均低于对照试剂检出限),对照试剂检出阴性。1例为考核试剂检出阴性,对照试剂检出阳性,可能原因为肿瘤组织异质性或检测方法学的差异。
考核试剂与对照试剂定性检测EGFR基因的阳性符合率为98.73%(95%CI 93.15%~99.97%),阴性符合率为96.45%(95%CI 91.92%~98.84%),总符合率为97.27%(95%CI 94.16%~98.99%)。进行Kappa一致性检验,Kappa值=0.941,P<0.001,显示二者具有良好的检测一致性。
考核试剂与对照试剂定性检测19Del的阳性符合率为95.00%(95%CI:83.08%~99.39%),阴性符合率为98.33%(95%CI:95.21%~99.65%),总符合率为97.73%(95%CI:94.78%~99.26%);EGFR L858R检测结果的阳性符合率为94.74%(95%CI:82.25%~99.36%),阴性符合率为98.35%(95%CI:95.26%~99.66%),总符合率为97.73%(95%CI:94.78%~99.26%);EGFR 稀有突变(包括T790M、L861Q、S768I等)检测结果的阳性符合率为100.00%(95%CI:54.07%~100.00%),阴性符合率为100.00%(95%CI:98.29%~100.00%),总符合率为100.00%(95%CI:98.34%~100.00%)。
1.2. ALK基因与已上市伴随诊断试剂盒的比较研究
ALK基因融合(重排)对比试剂采用雅培公司VysisALK Break Apart FISH 探针试剂盒和罗氏公司VENTANA ALK免疫组化(IHC)检测试剂盒。本次对比研究共纳入228例福尔马林固定,石蜡包埋的晚期非小细胞肺癌组织样本。228例样本的FISH与IHC检测结果有4例不一致,224例一致。考核试剂在224例样本中共检出29例阳性。
考核试剂在224例样本中有2例样本检测结果与对照试剂不一致。1例为考核试剂检出阳性(其测序丰度较低),对照试剂(IHC/FISH)均检出阴性。1例为考核试剂检出阴性,对照试剂(IHC/FISH)均检出阳性,可能原因为肿瘤组织异质性或检测方法学的差异。
考核试剂与对照试剂(IHC/FISH)定性检测ALK基因的阳性符合率为96.55%(95%CI:82.24%~99.91%),阴性符合率为99.49%(95%CI:97.18%~99.99%),总符合率为99.11%(95%CI:96.81%~99.89%)。进行Kappa一致性检验,Kappa值=0.960,P<0.001,显示二者具有良好的检测一致性。
1.3. ROS1基因与已上市伴随诊断试剂盒的比较研究
ROS1基因融合(重排)对比试剂采用厦门艾德生物人类ROS1基因融合检测试剂盒(荧光PCR法)。本次对比研究共纳入232例福尔马林固定,石蜡包埋的晚期非小细胞肺癌组织样本。考核试剂在7例样本中检出阳性。
考核试剂在232例样本中有1例样本检测结果与对照试剂不一致。可能原因为肿瘤组织异质性或检测方法学的差异。
考核试剂与对照试剂定性检测ROS1基因的阳性符合率为87.50%(95%CI 47.35%~99.68%),阴性符合率为100.00%(95%CI 98.37%~100.00%),总符合率为99.57%(95%CI 97.62%~99.99%)。进行Kappa一致性检验,Kappa值=0.931,P<0.001,显示二者具有良好的检测一致性。
1.4.KRAS基因与已上市伴随诊断试剂盒的比较研究
KRAS基因突变对比试剂采用凯杰公司therascreen®KRAS RGQ PCR Kit试剂盒。本次对比研究共纳入204例福尔马林固定,石蜡包埋的晚期结直肠癌组织样本。考核试剂在68例样本中检出阳性。
考核试剂在204例样本中有5例样本检测结果与对照试剂不一致。5例不一致样本均为考核试剂检出阳性(其测序丰度均低于对照试剂检测限),对照试剂检出阴性。
考核试剂与对照试剂定性检测KRAS基因的阳性符合率为 100.00%(95%CI:94.31%~100.00%),阴性符合率为96.45%%(95%CI:91.92%~98.84%),总符合率为97.55%(95%CI:94.37%~99.20%)。进行Kappa一致性检验,Kappa值=0.944,P<0.001,显示二者具有良好的检测一致性。
(三)TKI 药物疗效相关的回顾性临床研究
申请人在上海市肺科医院、北京胸科医院和山西省肿瘤医院进行靶向药物疗效相关的回顾性临床研究。3家研究中心合计完成162例靶向药物疗效相关研究。其中吉非替尼片53例、盐酸埃克替尼片6例、盐酸厄洛替尼片2例、甲磺酸奥西替尼片25例、克唑替尼胶囊53例、西妥昔单抗注射液23例。
1.吉非替尼片药效相关性研究
合计纳入53例既往接受吉非替尼片治疗的局部晚期或转移性非小细胞肺癌患者进行回顾性疗效分析。考核试剂在53名患者治疗前的石蜡包埋组织样本中均检出EGFR敏感突变阳性,与临床既往分子检测结果一致。在53名患者吉非替尼片疗效评估中,38例评估部分缓解,14例评估疾病稳定,1例评估疾病进展,临床用药客观缓解率为71.70%(95%CI 57.65%~83.21%),疾病控制率为98.11%(95%CI 89.93%~99.95%),与既往药物临床试验客观缓解率范围基本相符。
2.甲磺酸奥希替尼片药效相关性研究
合计纳入25例既往接受甲磺酸奥希替尼片治疗的局部晚期或转移性非小细胞肺癌患者进行回顾性疗效分析。考核试剂在25名患者治疗前的石蜡包埋组织样本中均检出EGFR基因T790M突变阳性,与临床既往分子检测结果一致。在25名患者甲磺酸奥希替尼片疗效评估中,16例评估部分缓解,8例评估疾病稳定,1例评估疾病进展,临床用药客观缓解率为64%(95%CI 42.52%~82.03%),疾病控制率为96%(95%CI 79.65%~99.90%),与既往药物临床试验客观缓解率范围基本相符。
3.克唑替尼胶囊(ALK融合)药效相关研究
合计纳入41例既往接受克唑替尼胶囊治疗的局部晚期或转移性非小细胞肺癌患者进行回顾性疗效分析。考核试剂在41名患者治疗前的石蜡包埋组织样本中均检出ALK基因重排(融合)阳性,与临床既往分子检测结果一致。在41名患者克唑替尼胶囊疗效评估中,30例评估部分缓解,9例评估疾病稳定,2例评估疾病进展,临床用药客观缓解率为73.17%(95%CI 57.06%~85.78%)、疾病控制率为95.12%(95%CI 83.47%~99.40%),与既往药物临床试验客观缓解率范围基本相符。
4.克唑替尼胶囊(ROS1融合)药效相关研究
合计纳入12例既往接受克唑替尼胶囊治疗的局部晚期或转移性非小细胞肺癌患者进行回顾性疗效分析。考核试剂在12名患者治疗前的石蜡包埋组织样本中均检出ROS1重排(融合)阳性,与临床既往分子检测结果一致。在12名患者克唑替尼胶囊疗效评估中,9例临床评估部分缓解,3例评估疾病稳定,临床用药客观缓解率为75%(95%CI 42.81%~94.51%)、疾病控制率为100%(95%CI 73.54%~100%),与既往药物临床试验客观缓解率范围基本相符。 5.西妥昔单抗注射液药效相关研究 合计纳入23例既往接受西妥昔单抗注射液治疗的晚期结直肠癌患者进行回顾性疗效分析。考核试剂在23名患者治疗前的石蜡包埋组织样本中的检测结果均为KRAS基因野生型,与临床既往分子检测结果一致。其中10名患者为初次治疗,与药物临床试验入组人群相符。在这10名患者西妥昔单抗注射液疗效评估中,7例评估部分缓解,2例评估疾病稳定,1例评估疾病进展,临床用药客观缓解率为70%(95%CI 34.75%~93.33%)、疾病控制率为90%(95%CI 55.50%~99.75%),与既往药物临床试验客观缓解率范围基本相符。另有11例为化疗后复发结直肠癌患者,治疗后疾病控制率为72.73%。其他2例为维持治疗。 综上所述,该产品临床试验资料对产品的临床性能进行了较全面研究,临床试验符合要求。
四、收益-风险评估
(一)收益评估
本产品用于定性检测非小细胞肺癌(NSCLC)、结直肠癌(CRC)患者经中性福尔马林固定的石蜡包埋(FFPE)的组织样本中EGFR/ALK/ROS1/RET/KRAS/NRAS/PIK3CA/BRAF/HER2/MET基因变异。其中,针对NSCLC,EGFR基因中:19号外显子缺失(19del)、L858R点突变用于吉非替尼片的伴随诊断检测,T790M点突变用于甲磺酸奥希替尼片的伴随诊断检测;ALK基因重排(融合)和ROS1基因重排(融合)用于克唑替尼胶囊的伴随诊断检测;针对CRC,KRAS基因野生型用于西妥昔单抗注射液的伴随诊断检测。
本试剂盒检测既适用于非小细胞肺癌(NSCLC)患者,亦适用于结直肠癌(CRC)患者。可一次性检测患者经中性福尔马林固定的石蜡包埋(FFPE)组织样本中的10种基因变异。患者通过检测结果可能受益于相应的靶向药物治疗,亦可能通过检测结果来辅助诊断,提示预后,获得更优的治疗方案。
(二)风险评估
本试剂盒检测结果会受到样本来源、样本采集过程、样本质量、样本运输条件、样本预处理等因素影响,同时也受到样本DNA提取质量、实验操作、实验环境等限制,导致可能得出假阳性或假阴性的检测结果。使用者须了解检测过程中可能存在的潜在风险及检测的局限性。不符合说明书中【样本要求】的样本和不恰当的实验操作会导致假阳性或假阴性结果,请严格按照产品说明书中【样本要求】及【检验方法】的要求操作和进行实验过程质控。同时由于肿瘤组织可能存在较大异质性,不同部位取样可能会得到不同的检测结果。本试剂盒阴性的检测结果不能完全排除靶基因突变的存在,样本中肿瘤细胞过少、过度降解、突变类型不在试剂盒检测范围内或靶基因浓度低于检测限亦可造成假阴性结果。 本试剂盒在检测过程中涉及基因扩增,在非可控的实验室操作可能由于环境中气溶胶的存在导致结果不可靠,同时PCR操作过程中气溶胶的泄露可能会导致设备甚至实验室的污染。因此,请在可控的实验室进行检测操作,操作人员需根据《医疗机构临床基因扩增管理办法》进行专业培训,非专业的操作及操作不当会影响检测质量。本试剂盒为基于二代测序平台的多基因位点的伴随诊断产品,检测过程主要包括FFPE样本中DNA提取、文库制备、杂交捕获、测序及数据分析等步骤。为确保检测全流程的质量得到有效控制,在产品设计开发阶段对样本提取试剂、文库质控试剂及测序试剂进行了全流程匹配性验证,为了确保检测结果的准确性,配套开发了测序数据分析及结果判读的生物信息分析软件。请使用本试剂盒推荐使用的配套试剂盒及软件进行检测,本试剂盒未对其他配套试剂盒及软件进行对比验证。
(三)其他因素
申报产品对临床疾病治疗的临床指导价值仍需要进一步临床研究。其它稀有突变本产品检测试剂可以检出,但相关肿瘤药物安全性和有效性尚未确定。
(四)收益-风险的确定
通过在说明书等文档中详细规定操作流程和注意事项、对实验人员进行专业培训等防范措施,对该产品的已知和可预见的安全风险进行控制和降低,剩余风险可以被控制在验收准则规定的可接受范围内,同时没有带来新的危害与安全风险。考虑到风险控制措施已明确而且申报产品不是判断临床疾病的唯一依据,临床中还应结合患者的临床病史、症状、其他诊断结果和临床医生的专业判断来综合评价临床疾病,在权衡获得的收益以及对临床风险的容忍度后,申报产品的收益大于风险。
综合评价意见
本申报项目为境内第三类医疗器械产品注册,属于创新审批项目(编号:201600077)。申请人的注册申报资料符合现行要求,依据《医疗器械监督管理条例》(国务院令第680号)、《体外诊断试剂注册管理办法》(国家食品药品监督管理总局令2014 年第5 号)等相关医疗器械法规与配套规章,经系统评价后,建议准予注册。申请人在该产品上市后应继续对产品伴随诊断用途进行验证。请在至少两家临床机构随访收集伴随诊断4 种药物(吉非替尼片、甲磺酸奥希替尼片、克唑替尼胶囊、西妥昔单抗注射液)的临床用药疗效随访数据,作为临床补充资料在产品下一次延续注册时提交。临床用药疗效随访数据应包括:病理诊断信息,应用本产品检测信息,患者用药疗效终点至最佳疗效的疗效数据,每种药物相关数据应满足统计学意义。该项临床资料应由出具数据的各临床试验机构签章。
2018年11月1日
© 2018 - 2020, Wuhan Tacro Technology Co.,Ltd All Rights Reserved.

